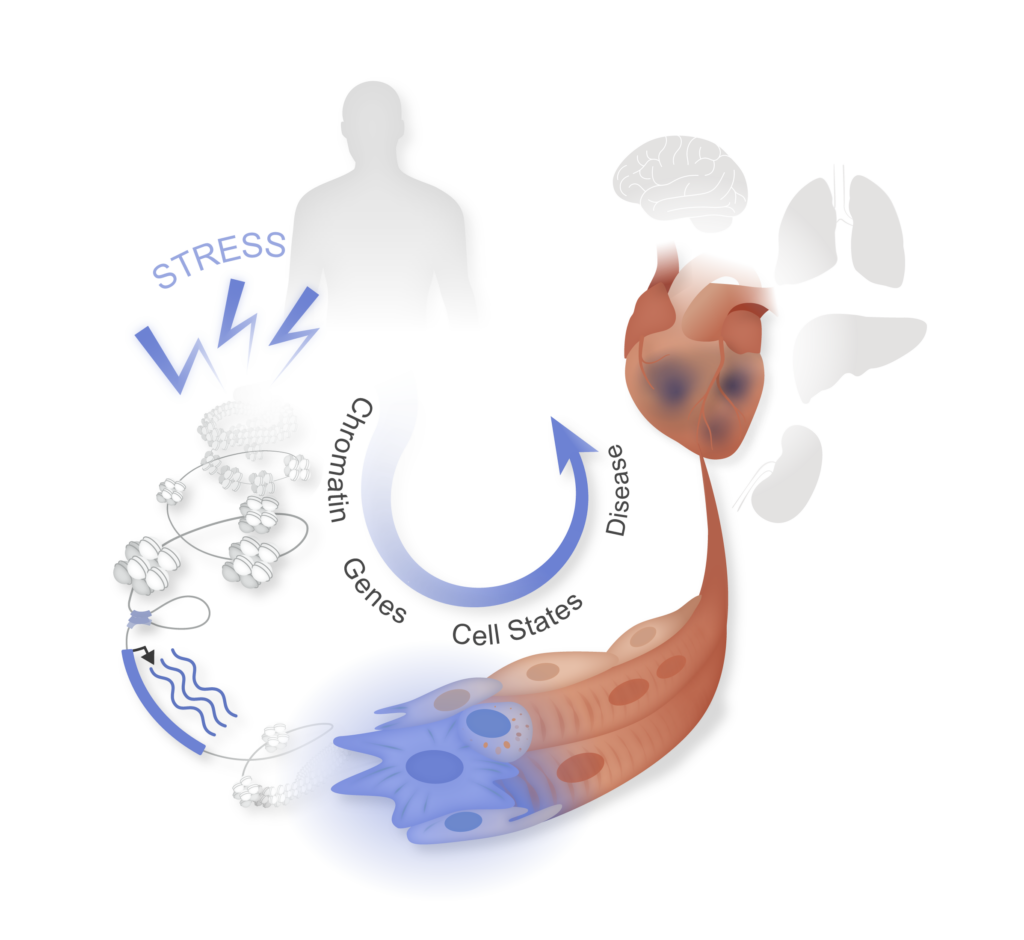
Heart disease is a global epidemic and accounts for 1 in every 5 deaths in the US. Currently available therapeutic modalities to treat cardiovascular disease such as heart failure do not directly alter root-cause defects in cardiac tissue and therefore only slow disease progression rather than preventing or reversing it. The fact that nearly half of those hospitalized with heart failure die within 5 years highlights the urgent need to identify completely new axes of disease pathogenesis and leverage this knowledge towards the development of new therapies.
Cardiovascular epigenomics
Targeting gene regulation has emerged as a new therapeutic strategy in a variety of chronic diseases. The central theme of our research program is to understand the gene-regulatory and epigenomic mechanisms that govern cellular plasticity in normal and abnormal physiology, with a particular focus on heart failure. Our main goal is to identify defined cis-elements, trans- factors and maladaptive cell states that can be targeted to treat maladaptive remodeling of tissue architecture in heart failure and other chronic diseases.
Our approaches
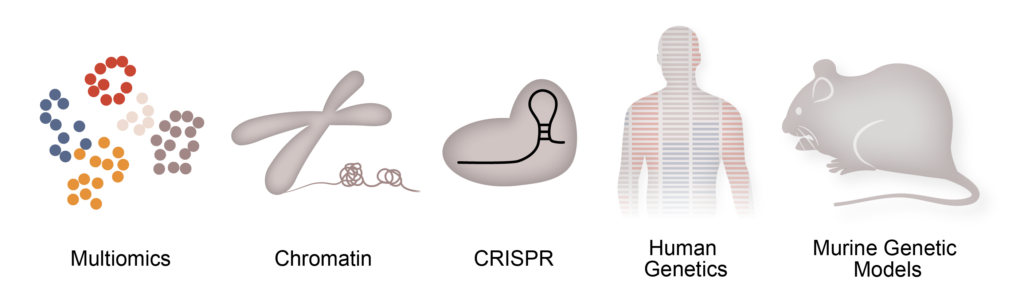
To achieve our goals we leverage a host of specialized techniques and tools to address these questions, including single cell genomics, assays to capture chromatin states (ChIP-seq, ATAC-seq, PRO-seq, CUT&RUN), genome editing via CRISPR-Cas9, human genetics, whole organ in vivo physiology, murine genetic models and computational biology.
Modulate fibroblast
plasticity in disease
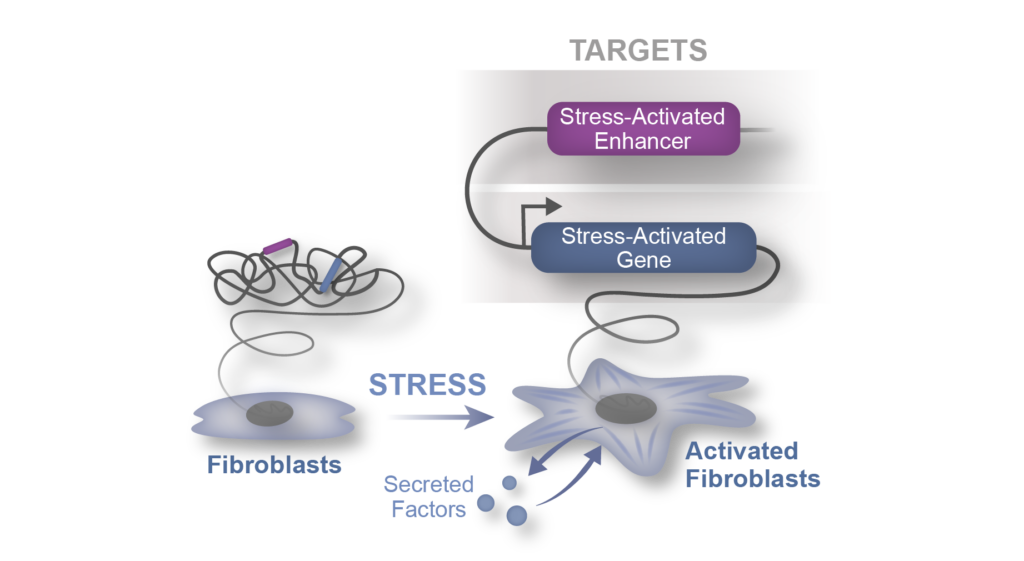
Fibroblast activation is a common stress response in tissues that worsens dysfunction of lung, liver, kidney, heart, and exacerbates many cancers, yet its mechanistic basis remains unclear. The stress-induced pathways that trigger fibroblast activation (e.g., TGFβ) ultimately converge on transcription factors and the chromatin regulatory apparatus in the nucleus, which transduce these broad upstream signals into changes in gene expression and cell identity. One of our goals is to discover new cellular and molecular mechanisms that govern the plasticity of fibroblast states in heart failure, findings which are applicable to a wide array of chronic diseases that feature tissue fibrosis (e.g., lung, liver and kidney).
Epigenomic regulation
of Inflammation
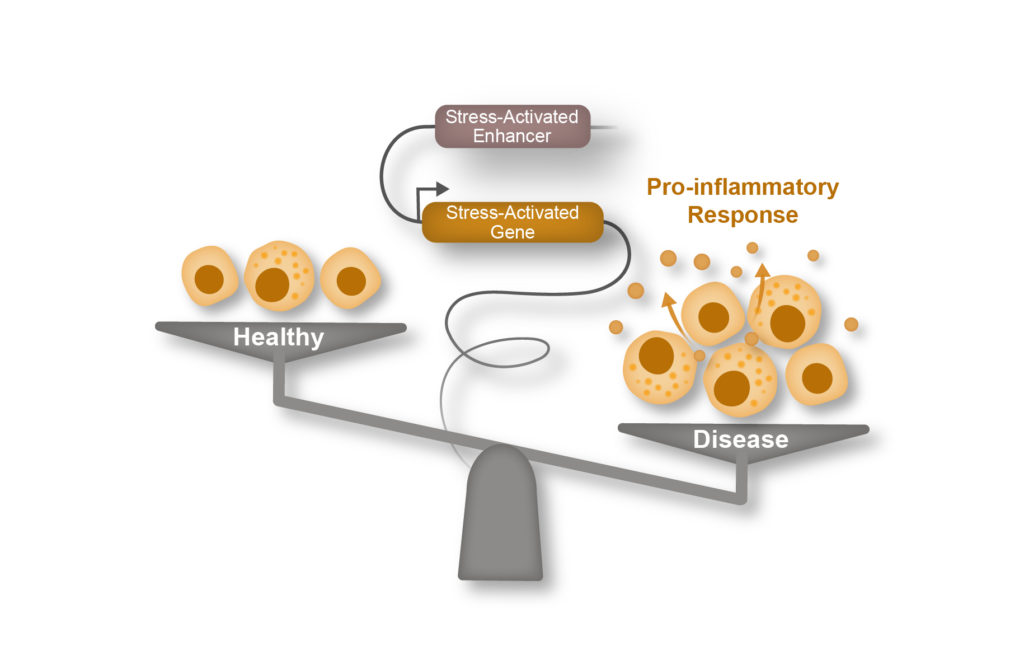
Over the past two decades it has become increasingly clear that innate and adaptive immune cell populations play a critical role in both heart homeostasis and disease. Our laboratory combines genomics with in vivo physiology and murine models of heart failure to understand the molecular mechanisms governing how chromatin and cell states are modulated in acute and chronic inflammation. We believe that such approaches can identify novel nodal regulators of the immune response in heart failure and other chronic diseases featuring inflammation.
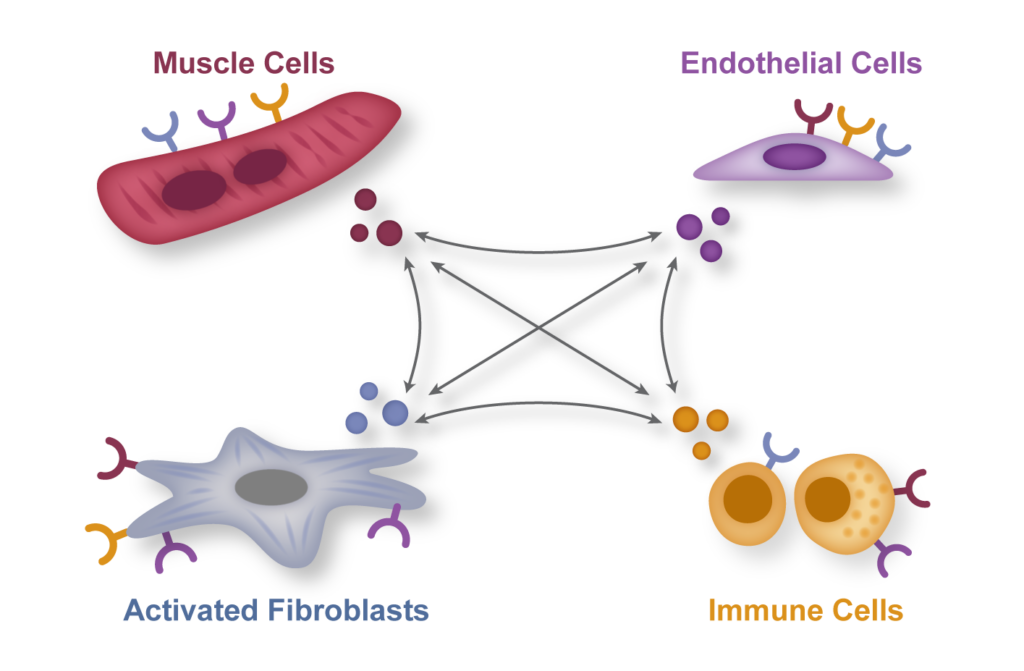
Cell-to-cell communication
The homeostasis of the heart, or that of any organ, relies on a complex cross-talk between different cell types with specialized functions. Recent advances in single cell omics have highlighted the heterogeneity of the cellular composition of the heart, revealing multiple subsets of cardiomyocytes, fibroblasts, endothelial and immune cells with diverse developmental origins and molecular properties. One of the goals of our lab is to decipher the molecular mechanisms governing pathological cross talk between diverse cellular compartments in heart disease.
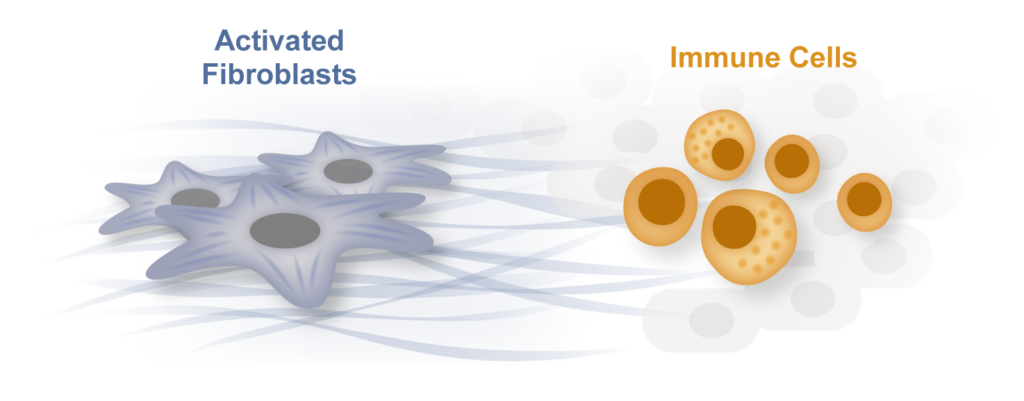
Inflammation and fibrosis: partners in heart disease and in many other human conditions
Inflammation and fibrosis are common stress responses that worsen organ function across a wide variety of human diseases, yet the molecular mechanisms governing their crosstalk are poorly understood. We aim to identify critical signaling molecules governing the crosstalk between the immune and fibroblast compartments in heart failure and explore if these mechanisms contribute to other diseases associated with inflammation and fibrosis in organs such as the lung, liver and the brain.
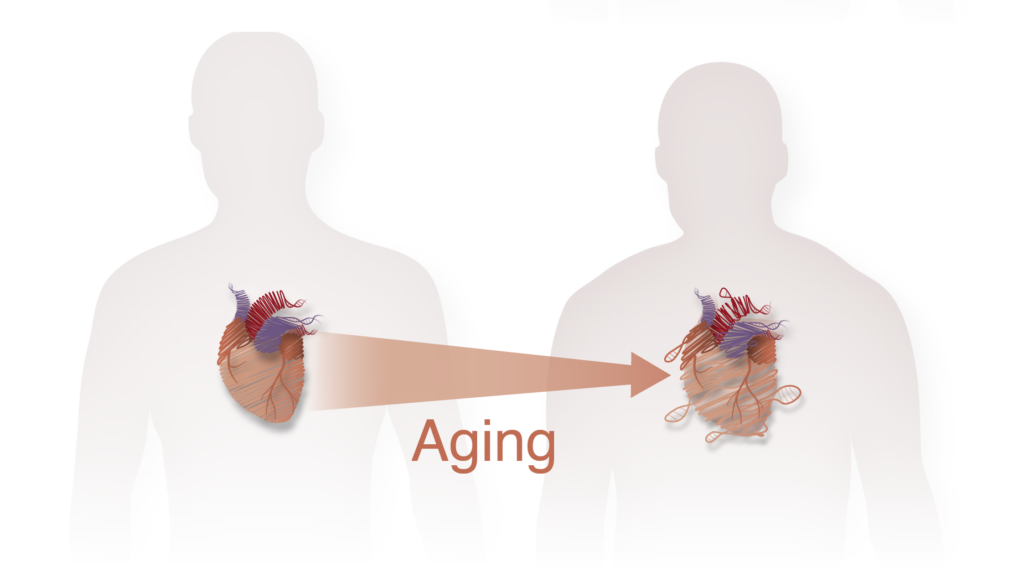
Epigenomic mechanisms of cardiac aging
As our average lifespan is continuously increasing, cardiovascular disease represents an even bigger threat for our societies. While much of the prior work in cardiac aging is focused on the role of mitochondria dysfunction and oxidative stress, very little is known about the transcriptional and epigenomic mechanisms underlying this process. We aim to identify and then perturb specific chromatin and cell states arising during aging in the cardiac tissue, with the goal of identifying novel molecular mechanisms governing this process and possible prevention and treatment methods.